17/02/2012
Molecular Orbital (MO) diagrams serve as profoundly insightful tools for comprehending the intricate nature of chemical bonding, offering a more nuanced perspective than simpler models like Lewis structures. They illustrate how atomic orbitals (AOs) from individual atoms combine to form new, molecule-wide orbitals. A common point of curiosity, however, arises when observing these diagrams: why are the original atomic orbitals not explicitly 'shown' as the final energy levels within the central part of the diagram? The answer lies in the fundamental principle of molecular orbital theory: atomic orbitals are the fundamental building blocks, the ingredients, but they undergo a profound transformation, ceasing to exist in their original isolated form. Instead, they merge to form entirely new, delocalised molecular orbitals that span the entire molecule. The MO diagram, therefore, represents the outcome of this transformation, with the AOs depicted on the periphery as the starting points, rather than as the final, internal states.
- The Atomic Foundation: What Are Atomic Orbitals?
- Building Molecules: The Linear Combination of Atomic Orbitals (LCAO) Approach
- The Rules of Engagement: Energy and Symmetry Matching
- A Deeper Insight: The Phenomenon of s-p Mixing
- Variations on a Theme: Homonuclear vs. Heteronuclear Diatomics
- Moving to Complexity: Triatomic Molecules
- The Ultimate Reason: MOs Are Not AOs
- Frequently Asked Questions (FAQs)
- Conclusion
The Atomic Foundation: What Are Atomic Orbitals?
To truly grasp why atomic orbitals are depicted as precursors rather than components within the molecular orbital energy landscape, one must first appreciate their nature. Atomic orbitals are quantum mechanical constructs, mathematically derived solutions to the Schrödinger equation for an isolated atom. Each AO describes a specific region around the atomic nucleus where an electron is most likely to be found, characterised by its energy, shape, and spatial orientation. Familiar examples include the spherical 1s and 2s orbitals, and the dumbbell-shaped 2p orbitals (2px, 2py, 2pz), each possessing distinct energy levels. In an isolated atom, electrons occupy these orbitals according to principles like the Aufbau principle, Hund's rule, and the Pauli exclusion principle. However, when atoms approach each other to form a chemical bond, these individual atomic identities begin to dissolve, paving the way for a more collective electron arrangement.
Building Molecules: The Linear Combination of Atomic Orbitals (LCAO) Approach
The cornerstone of molecular orbital theory is the Linear Combination of Atomic Orbitals (LCAO) approximation. This mathematical approach posits that molecular orbitals are formed by the algebraic addition and subtraction of atomic orbitals belonging to the constituent atoms. When AOs combine, they can do so in several ways, leading to different types of molecular orbitals:
- Bonding Molecular Orbitals: These arise from the constructive interference of atomic orbitals. The electron density is increased in the region between the nuclei, leading to a lower energy state and thus stabilising the bond. This increased electron density acts as a 'glue' holding the atoms together.
- Antibonding Molecular Orbitals: Formed by the destructive interference of atomic orbitals. Here, a nodal plane exists between the nuclei, meaning electron density is reduced to zero in this crucial bonding region. This leads to a higher energy state compared to the original AOs, destabilising the bond. Electrons in antibonding orbitals weaken the bond.
- Non-bonding Molecular Orbitals: In some cases, atomic orbitals may not interact effectively due to differences in symmetry or energy. These AOs then form non-bonding MOs, which essentially remain localised on a single atom, behaving much like lone pairs and having little to no effect on the overall bond strength.
The critical insight here is that once AOs combine, they cease to exist as discrete entities within the molecule. Instead, their electron clouds merge, resulting in new, delocalised orbitals that encompass the entire molecular framework. The MO diagram visually represents these newly formed energy levels, providing a comprehensive picture of how electrons are distributed throughout the molecule, rather than merely within individual atomic spheres.
The Rules of Engagement: Energy and Symmetry Matching
The formation of molecular orbitals from atomic orbitals is not a random process; it is governed by two crucial quantum mechanical rules: energy proximity and symmetry compatibility. For significant interaction and effective mixing to occur between atomic orbitals, they must adhere to these criteria.
- Energy Proximity: Atomic orbitals must have comparable energies to mix effectively. If the energy difference between two interacting AOs is substantial, their overlap will be negligible, and they will form either very weak bonds or simply remain as non-bonding orbitals. For instance, a 1s orbital from one atom will typically not interact strongly with a 2p orbital from another atom if their energy levels are vastly different. This principle becomes particularly important in heteronuclear molecules, where the differing electronegativities of the atoms lead to inherent energy differences in their respective atomic orbitals.
- Symmetry Compatibility: Atomic orbitals must possess the correct spatial symmetry with respect to the molecular axis or plane to overlap constructively. For example, a 2pz orbital (oriented along the internuclear axis) can form a sigma (σ) bond by head-on overlap with another 2pz orbital or an s orbital. Conversely, 2px and 2py orbitals, oriented perpendicular to the internuclear axis, can overlap side-on to form pi (π) bonds. Orbitals with mismatched symmetries, even if energetically close, cannot interact to form molecular orbitals because their overlaps would cancel out. The concept of symmetry becomes even more critical when analysing polyatomic molecules, where group theory is often employed to classify atomic and molecular orbitals based on the molecule's overall symmetry elements.
These rules dictate which AOs contribute to which MOs, and ultimately, the shape and energy of the resultant molecular orbitals. Without meeting these conditions, the transformation from atomic to molecular orbitals simply doesn't occur, or it occurs to a very limited, insignificant extent.
A Deeper Insight: The Phenomenon of s-p Mixing
One of the most fascinating aspects of molecular orbital theory, which further highlights the dynamic nature of orbital interactions, is the phenomenon of s-p mixing. This occurs when molecular orbitals of the same symmetry, formed from the initial combination of 2s and 2p atomic orbitals, are sufficiently close in energy to interact further. This secondary interaction can lead to a significant change in the expected order of orbital energies, profoundly impacting the electronic configuration and properties of a molecule.
Traditionally, in diatomic molecules, one might expect the 3σg MO (primarily derived from 2pz AOs) to be lower in energy than the 1πu MO (derived from 2px and 2py AOs). This order holds true for heavier second-row diatomics like O2 and F2. However, experimental observations and sophisticated computational models for lighter homonuclear diatomics, specifically from Li2 to N2, and certain heteronuclear combinations like CO and NO, reveal a different energy order: the 3σg MO is found to be higher in energy than (and therefore filled after) the 1πu MO.
This reversal is a direct consequence of s-p mixing. The 2σg bonding MO, primarily formed from the 2s atomic orbitals, and the 3σg MO, primarily from the 2pz atomic orbitals, both possess the same sigma (σ) symmetry and are 'gerade' (g). In lighter elements, the energy difference between the 2s and 2p atomic orbitals is smaller, making the 2σg and 3σg molecular orbitals energetically close enough to interact. This secondary interaction causes the lower-energy 2σg MO to be pushed even lower in energy, whilst the higher-energy 3σg MO is pushed significantly higher. For molecules like N2, this upward shift of the 3σg MO places it above the 1πu MO, leading to the observed orbital energy reversal.
To illustrate the energy levels, consider the approximate atomic orbital and molecular orbital energies (in Hartrees) for Nitrogen (N2) and Oxygen (O2) as examples:
| Orbital | N2 Energy (Hartrees) | O2 Energy (Hartrees) |
|---|---|---|
| 1s (AO) | -15.6289 | -20.6686 |
| 2s (AO) | -0.9452 | -1.2443 |
| 2p (AO) | -0.5677 | -0.6319 |
| 1σg | -15.6820 | -20.7296 |
| 1σu* | -15.6783 | -20.7286 |
| 2σg | -1.4736 | -1.6488 |
| 2σu* | -1.0987 | -1.4997 |
| 1πu | -0.6154 | -0.7052 |
| 3σg | -0.7504 | -0.6350 |
| 1πg* | N/A | -0.5319 |
Notice for N2, the 3σg (-0.7504) is higher than 1πu (-0.6154), while for O2, the 3σg (-0.6350) is lower than 1πu (-0.7052). This reversal is the hallmark of s-p mixing. Such phenomena underscore that molecular orbitals are not simple, static combinations but dynamic entities shaped by complex interactions.
Variations on a Theme: Homonuclear vs. Heteronuclear Diatomics
The principles of MO diagram construction remain consistent, but their application yields distinct results depending on whether the molecule is homonuclear (composed of two identical atoms) or heteronuclear (composed of two different atoms). These differences are primarily driven by variations in electronegativity.
- Homonuclear Diatomics: In molecules like H2, Li2, and N2, the two identical atoms contribute atomic orbitals of identical energy. This symmetry results in a balanced contribution from each atom to the molecular orbitals. For example, in N2, the s-p mixing effect is pronounced, leading to the 3σg orbital being higher in energy than the 1πu orbital. In O2 and F2, the energy difference between 2s and 2p AOs is larger, reducing the extent of s-p mixing, and thus the 'expected' order (3σg below 1πu) is observed. The MO diagram for O2 also famously explains its paramagnetic nature due to two unpaired electrons in the degenerate 1πg* antibonding orbitals.
- Heteronuclear Diatomics: When two different atoms combine, their atomic orbitals no longer have identical energy levels. The atomic orbitals of the more electronegative atom will be lower in energy than those of the less electronegative atom. This energy difference means that the molecular orbitals will be skewed, with greater contributions from the lower-energy AOs of the more electronegative atom. The 'g' and 'u' subscripts (gerade and ungerade), which denote symmetry with respect to an inversion centre, are no longer applicable as heteronuclear molecules lack this centre of symmetry.
Consider a few examples:
- Carbon Monoxide (CO): Isoelectronic with N2, CO shares similarities in its MO configuration (1σ21σ*22σ22σ*21π43σ2). However, due to oxygen being more electronegative than carbon, its 2s orbital is significantly lower in energy than carbon's 2s orbital. Consequently, the degree of s-p mixing is reduced, and the bonding MOs are polarised towards the oxygen atom.
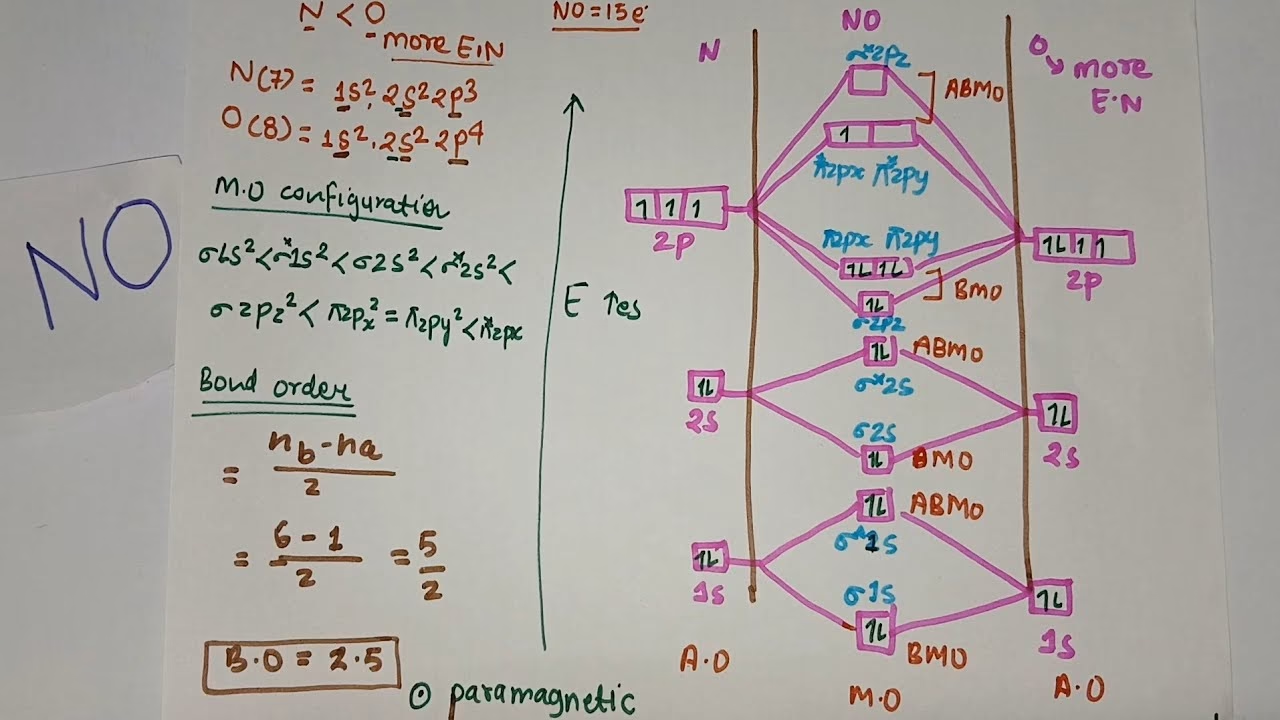
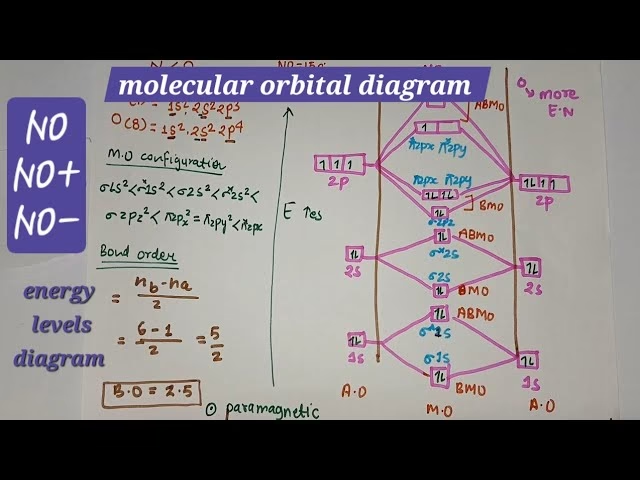
- Nitric Oxide (NO): This heteronuclear molecule also exhibits s-p mixing and is paramagnetic, possessing an unpaired electron. Its MO diagram shows that the energy differences between the 2s orbitals of nitrogen and oxygen are sufficient for each to largely form its own non-bonding σ orbitals, while the 2p orbitals mix to form bonding and antibonding π and σ orbitals. The bond order is 2.5, which is consistent with its stability and paramagnetic behaviour.
- Hydrogen Fluoride (HF): In HF, the hydrogen 1s orbital interacts primarily with the fluorine 2pz orbital (which is comparable in energy to the H 1s orbital) to form a sigma bond. The other fluorine 2p orbitals (2px and 2py) are non-bonding, existing as lone pairs, along with the fluorine 2s orbital. The HF electron configuration 1σ22σ23σ21π4 reflects this, resulting in a bond order of 1 and a diamagnetic molecule. The greater electronegativity of fluorine means its atomic orbitals are at lower energies, and the resulting bonding MOs are heavily weighted towards the fluorine atom, accounting for the polarity of the bond.
These examples underscore that MO diagrams provide a far more dynamic and accurate picture of electron distribution and bonding than static Lewis structures, incorporating the influence of atomic orbital energies and electronegativity differences.
Moving to Complexity: Triatomic Molecules
While diatomic molecules provide an excellent foundation, the principles of MO theory extend seamlessly to polyatomic molecules, albeit with increased complexity. For molecules with three or more atoms, the direct combination of individual atomic orbitals becomes cumbersome. Instead, a more sophisticated approach often involves forming 'group orbitals' or 'fragment orbitals' from symmetry-equivalent AOs, which then combine to form the molecular orbitals of the entire molecule. Here, the role of molecular symmetry becomes paramount, often requiring the application of group theory to classify and combine orbitals effectively.
- Carbon Dioxide (CO2): This is a linear molecule (D∞h symmetry). With carbon as the central atom, its 2s and 2p AOs combine with the 2s and 2p AOs of the two oxygen atoms. The oxygen 2s orbitals are significantly lower in energy than carbon's valence orbitals (O 2s at -32.4 eV vs. C 2s at -19.4 eV), meaning they remain largely non-bonding. The mixing occurs primarily between carbon's valence orbitals and oxygen's 2p orbitals. Symmetry labels (e.g., σ, π, g, u) are assigned based on the behaviour of the orbitals under the molecule's symmetry operations. The MO diagram for CO2 shows a rich set of bonding, non-bonding (from oxygen lone pairs), and antibonding orbitals, describing its double bonds and linearity.
- Water (H2O): Water is a bent molecule with C2v molecular symmetry. Its MO diagram is significantly different from linear molecules. The orbital symmetries are classified as a1, a2, b1, and b2, based on how they transform under the C2v symmetry operations (rotation, reflections). The oxygen atomic orbitals (2s, 2px, 2py, 2pz) are assigned corresponding symmetries. Crucially, the two hydrogen 1s orbitals first combine to form 'group orbitals' (one bonding, σ, and one antibonding, σ*), which then interact with the oxygen AOs of compatible symmetry and comparable energy.
For example, in water:
- The oxygen 2s (a1) mixes with the hydrogen σ group orbital (a1) to form the 2a1 MO.
- The oxygen 2py (b2) mixes with the hydrogen σ* group orbital (b2) to form the 1b2 MO.
- The oxygen 2pz (a1) mixes with other a1 orbitals (including some contribution from the 2s and hydrogen σ) to form the 3a1 MO.
- The oxygen 2px (b1), which is perpendicular to the molecular plane, does not have a hydrogen group orbital of compatible symmetry to interact with significantly, and thus forms a non-bonding 1b1 MO, essentially a lone pair.
This detailed MO treatment of water elegantly explains its photoelectron spectrum and provides a more accurate representation of its electron density than the traditional 'rabbit ear' lone pair model, revealing that the lone pairs are not entirely equivalent and are delocalised to some extent. The contrast with hydrogen sulphide (H2S), which also has C2v symmetry but a smaller bond angle, further illustrates how subtle changes in geometry and atomic orbital energies lead to distinct MO characteristics.
The Ultimate Reason: MOs Are Not AOs
In essence, the reason atomic orbitals are not 'shown' as distinct entities within the central energy levels of a molecular orbital diagram is fundamental: they have ceased to exist in their original form. AOs are theoretical constructs describing electrons in isolated atoms. When atoms bond, their electron wave functions overlap and combine, forming entirely new quantum mechanical states – the molecular orbitals. These MOs are delocalised, meaning the electrons occupying them are no longer confined to a single atom but are spread across the entire molecular framework. The MO diagram, therefore, is not a mapping of AOs onto a molecule; it is a representation of the *new*, integrated electronic structure of the molecule itself. The AOs are merely the basis set, the starting point from which these new molecular orbitals are constructed via the LCAO method. The diagram visually depicts this transformation, illustrating how the energy levels of the original atomic orbitals give rise to the new, often more stable, energy levels of the molecular orbitals.
Frequently Asked Questions (FAQs)
Q: Are atomic orbitals completely gone when molecular orbitals form?
A: Yes, in a sense, their individual identities are superseded. When atoms bond, their atomic orbitals combine and merge to form new molecular orbitals that span the entire molecule. The original AOs serve as the mathematical building blocks (a basis set) from which the MOs are constructed, but they do not remain as distinct, independent entities within the bonded molecule.
Q: Why are some orbitals non-bonding?
A: Non-bonding molecular orbitals form when atomic orbitals, or combinations thereof, do not have the appropriate symmetry or are too far apart in energy to interact effectively with other orbitals in the molecule. These orbitals essentially remain localised on a single atom, acting much like lone pairs of electrons and contributing little to the overall bonding or antibonding character of the molecule.
Q: How do we determine the exact energy order of molecular orbitals?
A: While the LCAO approximation provides a qualitative understanding, the precise energy order and values of molecular orbitals are determined through advanced quantum mechanical calculations, such as the Hartree-Fock method or Density Functional Theory (DFT). These computational methods provide quantitative solutions to the Schrödinger equation for molecules. Experimental techniques, notably photoelectron spectroscopy (PES), can also directly measure the ionisation energies of electrons from different molecular orbitals, thereby confirming their relative energy levels.
Q: Do all molecules exhibit s-p mixing?
A: Significant s-p mixing is most prominently observed in lighter homonuclear diatomic molecules (from Li2 to N2) and certain heteronuclear molecules (like CO and NO) where the energy difference between the 2s and 2p atomic orbitals is relatively small. This allows the 2s-derived and 2p-derived molecular orbitals of the same symmetry to interact and perturb each other's energies. In heavier elements, the larger energy gap between 2s and 2p AOs typically makes s-p mixing less significant or negligible.
Conclusion
Molecular Orbital theory offers a powerful and sophisticated framework for understanding the intricate world of chemical bonding, moving far beyond the simplistic representations of shared electron pairs. It reveals that the electronic structure of a molecule is not merely a collection of isolated atomic contributions but a new, integrated system of delocalised molecular orbitals. The depiction of atomic orbitals on the 'sides' of an MO diagram is thus not an oversight but a deliberate representation of their role as the fundamental precursors that undergo a profound transformation. By combining according to strict rules of energy and symmetry, atomic orbitals give rise to bonding, antibonding, and non-bonding molecular orbitals, which dictate a molecule's stability, magnetic properties, and reactivity. Understanding this transformation is key to appreciating the true quantum mechanical nature of chemical bonds.
If you want to read more articles similar to Unpacking Molecular Orbital Diagrams: The AO Transformation, you can visit the Automotive category.